Trait-related Trans-regulation Patterns
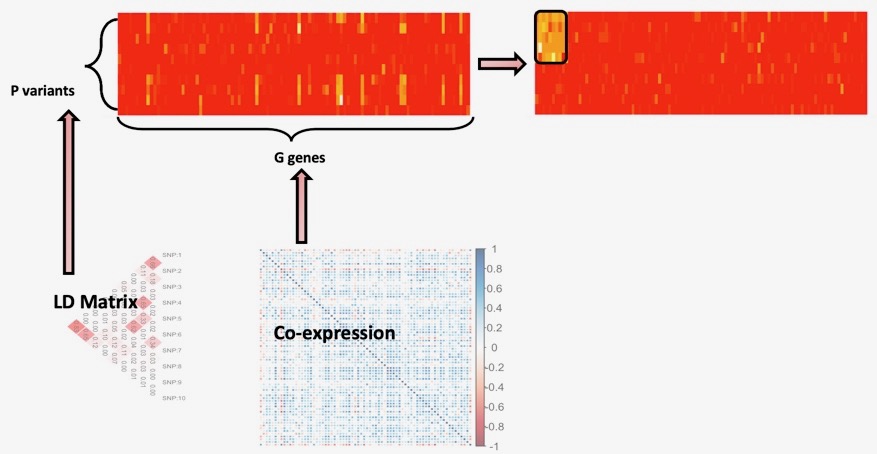
Trans-regulation of molecular phenotypes like gene-expression levels, protein levels, metabolites and others can provide key insights into causal mechanisms and pathways underlying complex diseases and traits. In general, trans-associations of variants associated to different traits, can highlight the key gene-sets and pathways involved in the genetic etiology of the trait in question. However, standard trans-mapping methods suffer from low statistical power due to high multiple testing burden and weaker effect sizes. To identify *trans*-targets of known trait-related variants, we employed a sparse canonical correlation based approach, named ARCHIE, which identifies gene-sets that are trans-regulated by sets of trait-related variants. This is particularly suitable for interpretation, since the identified gene-sets would represent major biological mechanisms via which the effects of the variants are cascaded. Applications of this method on summary statistics from trans-eQTL mapping in eQTLGen consortium, showed that the genes identified were indeed enriched in relevant pathways, transcription factor targets and trans-heritability. This approach, is further generalizable to any molecular phenotypes to detect broad regulation patterns. We are currently applying such methods to detect trans-regulation of plasma proteins by trait-related variants, regulation of gene-expression by copy number aberrations and their effect on cancer-related outcomes. With further generalizations into tensor decompositions and/or factor analysis, this can broadly be extend into multi-view data which will be critical in analyzing multi-omics data. See more in Publications.
![]()